Simulate spatial transcriptomic data
Dongyuan Song
Bioinformatics IDP, University of California, Los Angelesdongyuansong@ucla.edu
Qingyang Wang
Department of Statistics, University of California, Los Angelesqw802@g.ucla.edu
15 September 2023
Source:../../scDesign3/code/vignettes/scDesign3-spatial-vignette.Rmd
scDesign3-spatial-vignette.Rmd
library(scDesign3)
library(SingleCellExperiment)
library(ggplot2)
library(dplyr)
library(viridis)
theme_set(theme_bw())Introduction
In this tutorial, we show how to use scDesign3 to simulate the single-cell spatial data.
Read in the reference data
The raw data is from the Seurat, which is a dataset generated with the Visium technology from 10x Genomics. We pre-select the top spatial variable genes.
example_sce <- readRDS((url("https://figshare.com/ndownloader/files/40582019")))
print(example_sce)
#> class: SingleCellExperiment
#> dim: 1000 2696
#> metadata(0):
#> assays(2): counts logcounts
#> rownames(1000): Calb2 Gng4 ... Fndc5 Gda
#> rowData names(0):
#> colnames(2696): AAACAAGTATCTCCCA-1 AAACACCAATAACTGC-1 ...
#> TTGTTTCACATCCAGG-1 TTGTTTCCATACAACT-1
#> colData names(12): orig.ident nCount_Spatial ... spatial2 cell_type
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):To save time, we subset the top 10 genes.
example_sce <- example_sce[1:10, ]Simulation
Then, we can use this spatial dataset to generate new data by setting the parameter mu_formula as a smooth terms for the spatial coordinates.
set.seed(123)
example_simu <- scdesign3(
sce = example_sce,
assay_use = "counts",
celltype = "cell_type",
pseudotime = NULL,
spatial = c("spatial1", "spatial2"),
other_covariates = NULL,
mu_formula = "s(spatial1, spatial2, bs = 'gp', k= 400)",
sigma_formula = "1",
family_use = "nb",
n_cores = 2,
usebam = FALSE,
corr_formula = "1",
copula = "gaussian",
DT = TRUE,
pseudo_obs = FALSE,
return_model = FALSE,
nonzerovar = FALSE,
parallelization = "pbmcapply"
)Then, we can create the SinglecellExperiment object using the synthetic count matrix and store the logcounts to the input and synthetic SinglecellExperiment objects.
simu_sce <- SingleCellExperiment(list(counts = example_simu$new_count), colData = example_simu$new_covariate)
logcounts(simu_sce) <- log1p(counts(simu_sce))Visualization
We reformat the reference data and the synthetic data to visualize a few genes’ expressions and the spatial locations.
VISIUM_dat_test <- data.frame(t(log1p(counts(example_sce)))) %>% as_tibble() %>% dplyr::mutate(X = colData(example_sce)$spatial1, Y = colData(example_sce)$spatial2) %>% tidyr::pivot_longer(-c("X", "Y"), names_to = "Gene", values_to = "Expression") %>% dplyr::mutate(Method = "Reference")
VISIUM_dat_scDesign3 <- data.frame(t(log1p(counts(simu_sce)))) %>% as_tibble() %>% dplyr::mutate(X = colData(simu_sce)$spatial1, Y = colData(simu_sce)$spatial2) %>% tidyr::pivot_longer(-c("X", "Y"), names_to = "Gene", values_to = "Expression") %>% dplyr::mutate(Method = "scDesign3")
VISIUM_dat <- bind_rows(VISIUM_dat_test, VISIUM_dat_scDesign3) %>% dplyr::mutate(Method = factor(Method, levels = c("Reference", "scDesign3")))
VISIUM_dat %>% filter(Gene %in% rownames(example_sce)[1:5]) %>% ggplot(aes(x = X, y = Y, color = Expression)) + geom_point(size = 0.5) + scale_colour_gradientn(colors = viridis_pal(option = "magma")(10), limits=c(0, 8)) + coord_fixed(ratio = 1) + facet_grid(Method ~ Gene )+ theme_gray()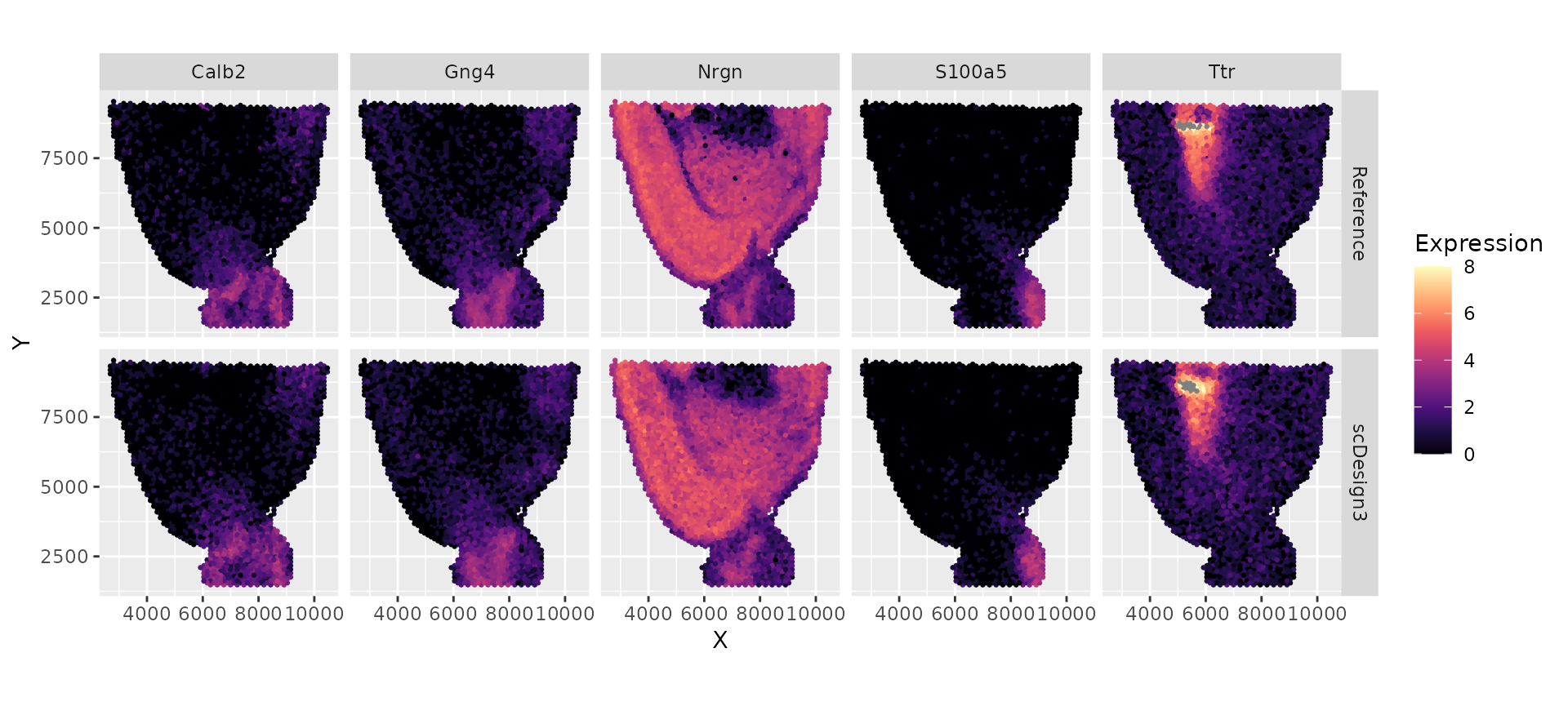
Session information
sessionInfo()
#> R version 4.3.1 (2023-06-16)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Ubuntu 20.04.6 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/liblapack.so.3; LAPACK version 3.9.0
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
#> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: America/Los_Angeles
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] viridis_0.6.4 viridisLite_0.4.2
#> [3] dplyr_1.1.2 ggplot2_3.4.2
#> [5] SingleCellExperiment_1.22.0 SummarizedExperiment_1.30.2
#> [7] Biobase_2.60.0 GenomicRanges_1.52.0
#> [9] GenomeInfoDb_1.36.1 IRanges_2.34.1
#> [11] S4Vectors_0.38.1 BiocGenerics_0.46.0
#> [13] MatrixGenerics_1.12.2 matrixStats_1.0.0
#> [15] scDesign3_0.99.6 BiocStyle_2.28.0
#>
#> loaded via a namespace (and not attached):
#> [1] gtable_0.3.3 xfun_0.39 bslib_0.5.0
#> [4] lattice_0.21-8 gamlss_5.4-12 vctrs_0.6.3
#> [7] tools_4.3.1 bitops_1.0-7 generics_0.1.3
#> [10] parallel_4.3.1 tibble_3.2.1 fansi_1.0.4
#> [13] highr_0.10 pkgconfig_2.0.3 Matrix_1.6-0
#> [16] desc_1.4.2 lifecycle_1.0.3 GenomeInfoDbData_1.2.10
#> [19] farver_2.1.1 compiler_4.3.1 stringr_1.5.0
#> [22] textshaping_0.3.6 munsell_0.5.0 htmltools_0.5.5
#> [25] sass_0.4.7 RCurl_1.98-1.12 yaml_2.3.7
#> [28] tidyr_1.3.0 crayon_1.5.2 pillar_1.9.0
#> [31] pkgdown_2.0.7 jquerylib_0.1.4 MASS_7.3-60
#> [34] DelayedArray_0.26.6 cachem_1.0.8 mclust_6.0.0
#> [37] nlme_3.1-162 tidyselect_1.2.0 digest_0.6.33
#> [40] mvtnorm_1.2-2 stringi_1.7.12 purrr_1.0.1
#> [43] bookdown_0.34 labeling_0.4.2 splines_4.3.1
#> [46] gamlss.dist_6.0-5 rprojroot_2.0.3 fastmap_1.1.1
#> [49] grid_4.3.1 gamlss.data_6.0-2 colorspace_2.1-0
#> [52] cli_3.6.1 magrittr_2.0.3 S4Arrays_1.0.4
#> [55] survival_3.5-5 utf8_1.2.3 withr_2.5.0
#> [58] scales_1.2.1 XVector_0.40.0 rmarkdown_2.23
#> [61] gridExtra_2.3 ragg_1.2.5 memoise_2.0.1
#> [64] evaluate_0.21 knitr_1.43 mgcv_1.9-0
#> [67] rlang_1.1.1 glue_1.6.2 BiocManager_1.30.21.1
#> [70] jsonlite_1.8.7 R6_2.5.1 zlibbioc_1.46.0
#> [73] systemfonts_1.0.4 fs_1.6.3