Simulate datasets with batch effect
Dongyuan Song
Bioinformatics IDP, University of California, Los Angelesdongyuansong@ucla.edu
Qingyang Wang
Department of Statistics, University of California, Los Angelesqw802@g.ucla.edu
15 September 2023
Source:../../scDesign3/code/vignettes/scDesign3-batchEffect-vignette.Rmd
scDesign3-batchEffect-vignette.RmdIntroduction
In this tutorial, we will show how to use scDesign3 to simulate data with original batch effects and how to remove the batch effects. We will also demostrate how to add ariticial batch effects.
Read in the reference data
The raw data is from the SeuratData package. The data is called pbmcsca in the package; it is PBMC Systematic Comparative Analysis dataset from the Broad Institute.
example_sce <- readRDS((url("https://figshare.com/ndownloader/files/40581965")))
print(example_sce)
#> class: SingleCellExperiment
#> dim: 1000 6276
#> metadata(0):
#> assays(2): counts logcounts
#> rownames(1000): LYZ GNLY ... PBXIP1 SECTM1
#> rowData names(0):
#> colnames(6276): pbmc2_10X_V2_AAACCTGAGATGGGTC
#> pbmc2_10X_V2_AAACCTGAGCGTAATA ... pbmc1_10x_v3_TTGAACGCATGCAGCC
#> pbmc1_10x_v3_TTTGACTAGTGTTCCA
#> colData names(13): phenoid orig.ident ... batch cell_type
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):To save computational time, we only use the top 100 genes.
example_sce <- example_sce[1:100, ]The column batch in this example dataset’s colData contains the batch information.
head(colData(example_sce))
#> DataFrame with 6 rows and 13 columns
#> phenoid orig.ident nCount_RNA
#> <character> <factor> <numeric>
#> pbmc2_10X_V2_AAACCTGAGATGGGTC B cell pbmc2 2360
#> pbmc2_10X_V2_AAACCTGAGCGTAATA B cell pbmc2 1888
#> pbmc2_10X_V2_AAACCTGAGCTAGGCA Cytotoxic T cell pbmc2 3456
#> pbmc2_10X_V2_AAACCTGAGGGTCTCC Dendritic cell pbmc2 3802
#> pbmc2_10X_V2_AAACCTGGTCCGAACC CD4+ T cell pbmc2 3826
#> pbmc2_10X_V2_AAACCTGTCGTCCGTT CD4+ T cell pbmc2 2345
#> nFeature_RNA nGene nUMI
#> <integer> <character> <character>
#> pbmc2_10X_V2_AAACCTGAGATGGGTC 1044 1044 2360
#> pbmc2_10X_V2_AAACCTGAGCGTAATA 803 803 1888
#> pbmc2_10X_V2_AAACCTGAGCTAGGCA 1372 1372 3456
#> pbmc2_10X_V2_AAACCTGAGGGTCTCC 1519 1519 3802
#> pbmc2_10X_V2_AAACCTGGTCCGAACC 1451 1451 3826
#> pbmc2_10X_V2_AAACCTGTCGTCCGTT 931 931 2345
#> percent.mito Cluster Experiment
#> <character> <character> <character>
#> pbmc2_10X_V2_AAACCTGAGATGGGTC 0.0419491525423729 2 pbmc2
#> pbmc2_10X_V2_AAACCTGAGCGTAATA 0.0413135593220339 2 pbmc2
#> pbmc2_10X_V2_AAACCTGAGCTAGGCA 0.0353009259259259 1 pbmc2
#> pbmc2_10X_V2_AAACCTGAGGGTCTCC 0.0420831141504471 6 pbmc2
#> pbmc2_10X_V2_AAACCTGGTCCGAACC 0.0371144798745426 0 pbmc2
#> pbmc2_10X_V2_AAACCTGTCGTCCGTT 0.0652452025586354 0 pbmc2
#> Method ident batch
#> <character> <factor> <character>
#> pbmc2_10X_V2_AAACCTGAGATGGGTC 10x Chromium (v2) pbmc2 10x Chromium (v2)
#> pbmc2_10X_V2_AAACCTGAGCGTAATA 10x Chromium (v2) pbmc2 10x Chromium (v2)
#> pbmc2_10X_V2_AAACCTGAGCTAGGCA 10x Chromium (v2) pbmc2 10x Chromium (v2)
#> pbmc2_10X_V2_AAACCTGAGGGTCTCC 10x Chromium (v2) pbmc2 10x Chromium (v2)
#> pbmc2_10X_V2_AAACCTGGTCCGAACC 10x Chromium (v2) pbmc2 10x Chromium (v2)
#> pbmc2_10X_V2_AAACCTGTCGTCCGTT 10x Chromium (v2) pbmc2 10x Chromium (v2)
#> cell_type
#> <character>
#> pbmc2_10X_V2_AAACCTGAGATGGGTC B cell
#> pbmc2_10X_V2_AAACCTGAGCGTAATA B cell
#> pbmc2_10X_V2_AAACCTGAGCTAGGCA Cytotoxic T cell
#> pbmc2_10X_V2_AAACCTGAGGGTCTCC Dendritic cell
#> pbmc2_10X_V2_AAACCTGGTCCGAACC CD4+ T cell
#> pbmc2_10X_V2_AAACCTGTCGTCCGTT CD4+ T cellSimulation
We can simulate a new data with batch effect information.
set.seed(123)
simu_res <- scdesign3(sce = example_sce,
assay_use = "counts",
celltype = "cell_type",
pseudotime = NULL,
spatial = NULL,
other_covariates = c("batch"),
mu_formula = "cell_type + batch",
sigma_formula = "1",
family_use = "nb",
n_cores = 2,
usebam = FALSE,
corr_formula = "1",
copula = "gaussian",
DT = TRUE,
pseudo_obs = FALSE,
return_model = FALSE)We can also remove the batch effect and generate new data.
BATCH_data <- construct_data(
sce = example_sce,
assay_use = "counts",
celltype = "cell_type",
pseudotime = NULL,
spatial = NULL,
other_covariates = c("batch"),
corr_by = "1"
)
BATCH_marginal <- fit_marginal(
data = BATCH_data,
predictor = "gene",
mu_formula = "cell_type + batch",
sigma_formula = "1",
family_use = "nb",
n_cores = 2,
usebam = FALSE
)
set.seed(123)
BATCH_copula <- fit_copula(
sce = example_sce,
assay_use = "counts",
marginal_list = BATCH_marginal,
family_use = "nb",
copula = "gaussian",
n_cores = 2,
input_data = BATCH_data$dat
)In here, we remove the batch effect by setting its coefficient to zero for all genes’ marginal fits. Then, we use the new sets of coefficients to generate the parameters for all genes across all cells.
BATCH_marginal_null <- lapply(BATCH_marginal, function(x) {
x$fit$coefficients[length(x$fit$coefficients)] <- 0
x
})
BATCH_para_null <- extract_para(
sce = example_sce,
marginal_list = BATCH_marginal_null,
n_cores = 2,
family_use = "nb",
new_covariate = BATCH_data$newCovariate,
data = BATCH_data$dat
)
set.seed(123)
BATCH_newcount_null <- simu_new(
sce = example_sce,
mean_mat = BATCH_para_null$mean_mat,
sigma_mat = BATCH_para_null$sigma_mat,
zero_mat = BATCH_para_null$zero_mat,
quantile_mat = NULL,
copula_list = BATCH_copula$copula_list,
n_cores = 2,
family_use = "nb",
input_data = BATCH_data$dat,
new_covariate = BATCH_data$newCovariate,
important_feature = BATCH_copula$important_feature,
filtered_gene = BATCH_data$filtered_gene
)Additionally, we can alter the batch effect information by mannually change the estimated coefficient for batch effect in each gene’s marginal model. Then, we can simulate new dataset with altered batch effect information.
BATCH_marginal_alter <- lapply(BATCH_marginal, function(x) {
x$fit$coefficients[length(x$fit$coefficients)] <- rnorm(1, mean = 5, sd = 2)
x
})
BATCH_para_alter <- extract_para(
sce = example_sce,
marginal_list = BATCH_marginal_alter,
n_cores = 2,
family_use = "nb",
new_covariate = BATCH_data$newCovariate,
data = BATCH_data$dat
)
set.seed(123)
BATCH_newcount_alter <- simu_new(
sce = example_sce,
mean_mat = BATCH_para_alter$mean_mat,
sigma_mat = BATCH_para_alter$sigma_mat,
zero_mat = BATCH_para_alter$zero_mat,
quantile_mat = NULL,
copula_list = BATCH_copula$copula_list,
n_cores = 2,
family_use = "nb",
input_data = BATCH_data$dat,
new_covariate = BATCH_data$newCovariate,
important_feature = BATCH_copula$important_feature,
filtered_gene = BATCH_data$filtered_gene
)We create three SingleCellExperiment objects using three count matrices generated above and store their logcounts.
Visulization
set.seed(123)
compare_figure <- plot_reduceddim(ref_sce = example_sce,
sce_list = simu_res_list,
name_vec = c("Reference", "w/ Batch", "w/o Batch","Aritifical Batch"),
assay_use = "logcounts",
if_plot = TRUE,
color_by = "cell_type",
shape_by = "batch",
n_pc = 20)
plot(compare_figure$p_umap)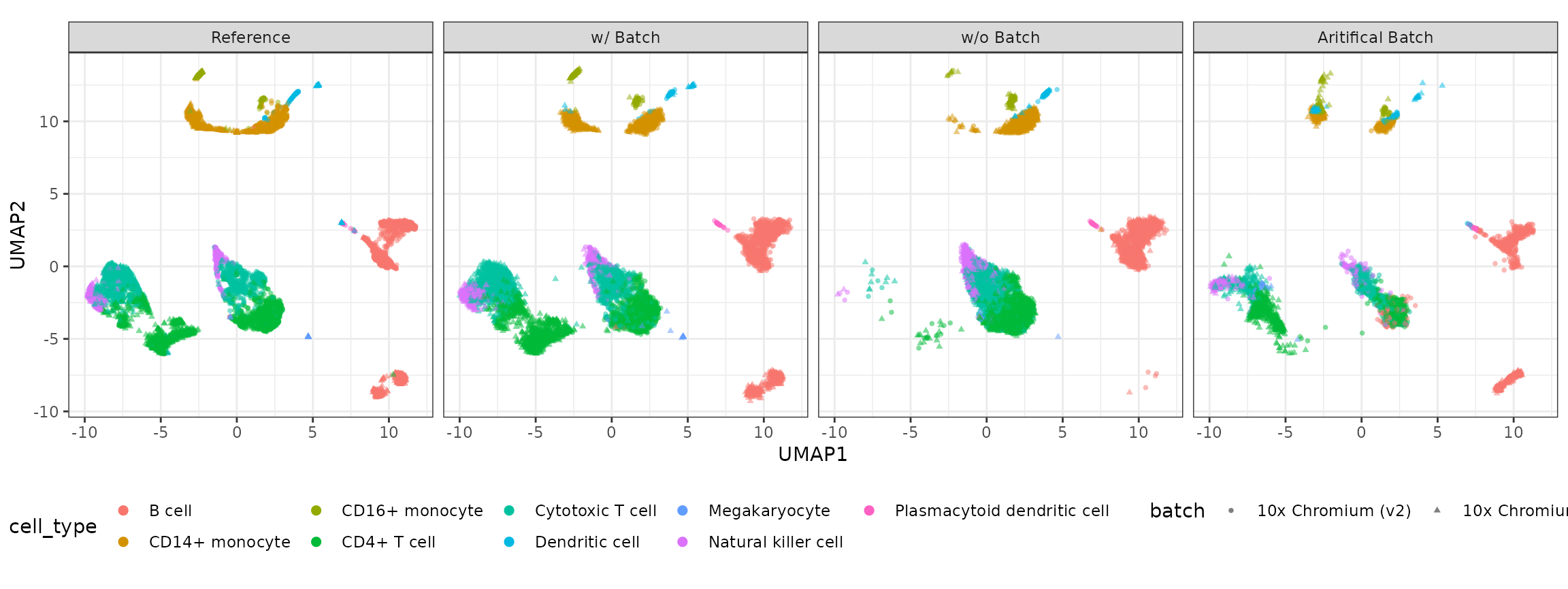
Session information
sessionInfo()
#> R version 4.3.1 (2023-06-16)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Ubuntu 20.04.6 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/liblapack.so.3; LAPACK version 3.9.0
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
#> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: America/Los_Angeles
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] SingleCellExperiment_1.22.0 SummarizedExperiment_1.30.2
#> [3] Biobase_2.60.0 GenomicRanges_1.52.0
#> [5] GenomeInfoDb_1.36.1 IRanges_2.34.1
#> [7] S4Vectors_0.38.1 BiocGenerics_0.46.0
#> [9] MatrixGenerics_1.12.2 matrixStats_1.0.0
#> [11] ggplot2_3.4.2 scDesign3_0.99.6
#> [13] BiocStyle_2.28.0
#>
#> loaded via a namespace (and not attached):
#> [1] tidyselect_1.2.0 farver_2.1.1 dplyr_1.1.2
#> [4] bitops_1.0-7 fastmap_1.1.1 RCurl_1.98-1.12
#> [7] digest_0.6.33 lifecycle_1.0.3 survival_3.5-5
#> [10] gamlss.dist_6.0-5 magrittr_2.0.3 compiler_4.3.1
#> [13] rlang_1.1.1 sass_0.4.7 tools_4.3.1
#> [16] utf8_1.2.3 yaml_2.3.7 knitr_1.43
#> [19] labeling_0.4.2 askpass_1.1 S4Arrays_1.0.4
#> [22] mclust_6.0.0 reticulate_1.30 DelayedArray_0.26.6
#> [25] withr_2.5.0 purrr_1.0.1 desc_1.4.2
#> [28] grid_4.3.1 fansi_1.0.4 colorspace_2.1-0
#> [31] scales_1.2.1 MASS_7.3-60 cli_3.6.1
#> [34] mvtnorm_1.2-2 rmarkdown_2.23 crayon_1.5.2
#> [37] ragg_1.2.5 generics_0.1.3 umap_0.2.10.0
#> [40] RSpectra_0.16-1 cachem_1.0.8 stringr_1.5.0
#> [43] zlibbioc_1.46.0 splines_4.3.1 parallel_4.3.1
#> [46] BiocManager_1.30.21.1 XVector_0.40.0 vctrs_0.6.3
#> [49] Matrix_1.6-0 jsonlite_1.8.7 bookdown_0.34
#> [52] gamlss_5.4-12 irlba_2.3.5.1 systemfonts_1.0.4
#> [55] jquerylib_0.1.4 glue_1.6.2 pkgdown_2.0.7
#> [58] stringi_1.7.12 gtable_0.3.3 munsell_0.5.0
#> [61] tibble_3.2.1 pillar_1.9.0 htmltools_0.5.5
#> [64] openssl_2.1.0 gamlss.data_6.0-2 GenomeInfoDbData_1.2.10
#> [67] R6_2.5.1 textshaping_0.3.6 rprojroot_2.0.3
#> [70] evaluate_0.21 lattice_0.21-8 highr_0.10
#> [73] png_0.1-8 memoise_2.0.1 bslib_0.5.0
#> [76] Rcpp_1.0.11 nlme_3.1-162 mgcv_1.9-0
#> [79] xfun_0.39 fs_1.6.3 pkgconfig_2.0.3