Simulate datasets with condition effect
Dongyuan Song
Bioinformatics IDP, University of California, Los Angelesdongyuansong@ucla.edu
Qingyang Wang
Department of Statistics, University of California, Los Angelesqw802@g.ucla.edu
15 September 2023
Source:../../scDesign3/code/vignettes/scDesign3-conditionEffect-vignette.Rmd
scDesign3-conditionEffect-vignette.RmdIntroduction
In this tutorial, we will show how to use scDesign3 to simulate data with condition effects and how to adjust the condition effects.
Read in the reference data
The raw data is from the SeuratData package. The data is called ifnb in the package; it is PBMC data simulated and controlled by IFNB.
example_sce <- readRDS((url("https://figshare.com/ndownloader/files/40581977")))
print(example_sce)
#> class: SingleCellExperiment
#> dim: 1000 13999
#> metadata(0):
#> assays(2): counts logcounts
#> rownames(1000): FTL HBB ... AC022182.3 SCIN
#> rowData names(0):
#> colnames(13999): AAACATACATTTCC.1 AAACATACCAGAAA.1 ... TTTGCATGCTAAGC.1
#> TTTGCATGGGACGA.1
#> colData names(8): orig.ident nCount_RNA ... cell_type condition
#> reducedDimNames(0):
#> mainExpName: RNA
#> altExpNames(0):
print(table(colData(example_sce)$cell_type))
#>
#> CD14 Mono CD4 Naive T CD4 Memory T CD16 Mono B CD8 T
#> 4362 2504 1762 1044 978 814
#> T activated NK DC B Activated Mk pDC
#> 633 619 472 388 236 132
#> Eryth
#> 55To save computational time, we only use the top 100 genes and two cell types (CD14 Mono and B).
example_sce <- example_sce[1:100, colData(example_sce)$cell_type %in% c("CD14 Mono", "B")]
## Remove unused cell type levels
colData(example_sce)$cell_type <- droplevels(colData(example_sce)$cell_type)The condition information is stored in colData of the example dataset.
head(colData(example_sce))
#> DataFrame with 6 rows and 8 columns
#> orig.ident nCount_RNA nFeature_RNA stim
#> <character> <numeric> <integer> <character>
#> AAACATACATTTCC.1 IMMUNE_CTRL 3017 877 CTRL
#> AAACATACCAGAAA.1 IMMUNE_CTRL 2481 713 CTRL
#> AAACATACCTCGCT.1 IMMUNE_CTRL 3420 850 CTRL
#> AAACATACGGCATT.1 IMMUNE_CTRL 1581 557 CTRL
#> AAACATTGCTTCGC.1 IMMUNE_CTRL 2536 669 CTRL
#> AAACGCACTCGCCT.1 IMMUNE_CTRL 3563 908 CTRL
#> seurat_annotations ident cell_type condition
#> <factor> <factor> <factor> <character>
#> AAACATACATTTCC.1 CD14 Mono IMMUNE_CTRL CD14 Mono CTRL
#> AAACATACCAGAAA.1 CD14 Mono IMMUNE_CTRL CD14 Mono CTRL
#> AAACATACCTCGCT.1 CD14 Mono IMMUNE_CTRL CD14 Mono CTRL
#> AAACATACGGCATT.1 CD14 Mono IMMUNE_CTRL CD14 Mono CTRL
#> AAACATTGCTTCGC.1 CD14 Mono IMMUNE_CTRL CD14 Mono CTRL
#> AAACGCACTCGCCT.1 CD14 Mono IMMUNE_CTRL CD14 Mono CTRLSimulation
First, we will simulate new data with the condition effects.
set.seed(123)
simu_res <- scdesign3(
sce = example_sce,
assay_use = "counts",
celltype = "cell_type",
pseudotime = NULL,
spatial = NULL,
other_covariates = c("condition"),
mu_formula = "cell_type + condition + cell_type*condition",
sigma_formula = "1",
family_use = "nb",
n_cores = 2,
usebam = FALSE,
corr_formula = "cell_type",
copula = "gaussian",
DT = TRUE,
pseudo_obs = FALSE,
return_model = FALSE,
nonzerovar = FALSE
)Then, we can also simulate a new dataset with condition effects on B cells removed.
IFNB_data <- construct_data(
sce = example_sce,
assay_use = "counts",
celltype = "cell_type",
pseudotime = NULL,
spatial = NULL,
other_covariates = c("condition"),
corr_by = "cell_type"
)
IFNB_marginal <- fit_marginal(
data = IFNB_data,
predictor = "gene",
mu_formula = "cell_type + condition + cell_type*condition",
sigma_formula = "1",
family_use = "nb",
n_cores = 2,
usebam = FALSE
)
set.seed(123)
IFNB_copula <- fit_copula(
sce = example_sce,
assay_use = "counts",
marginal_list = IFNB_marginal,
family_use = "nb",
copula = "gaussian",
n_cores = 2,
input_data = IFNB_data$dat
)In here, the condition effects on B cells are removed for all genes by modifying the estimated coefficients for all genes’ marginal models.
IFNB_marginal_null_B <- lapply(IFNB_marginal, function(x) {
x$fit$coefficients["cell_typeB:conditionSTIM"] <- 0-x$fit$coefficients["conditionSTIM"]
x
})Then, we can generate the parameters using the altered marginal fits and simulate new data with the altered paremeters.
IFNB_para_null_B <- extract_para(
sce = example_sce,
marginal_list = IFNB_marginal_null_B,
n_cores = 2,
family_use = "nb",
new_covariate = IFNB_data$newCovariate,
data = IFNB_data$dat
)
set.seed(123)
IFNB_newcount_null_B <- simu_new(
sce = example_sce,
mean_mat = IFNB_para_null_B$mean_mat,
sigma_mat = IFNB_para_null_B$sigma_mat,
zero_mat = IFNB_para_null_B$zero_mat,
quantile_mat = NULL,
copula_list = IFNB_copula$copula_list,
n_cores = 2,
family_use = "nb",
input_data = IFNB_data$dat,
new_covariate = IFNB_data$newCovariate,
important_feature = IFNB_copula$important_feature,
filtered_gene = IFNB_data$filtered_gene
)Then, we can create the SinglecellExperiment object using the synthetic count matrix and store the logcounts to the input and synthetic SinglecellExperiment objects.
Visulization
set.seed(123)
compare_figure <- plot_reduceddim(ref_sce = example_sce,
sce_list = simu_res_list,
name_vec = c("Reference", "w/ Condition", "Modified Condition"),
assay_use = "logcounts",
if_plot = TRUE,
color_by = "cell_type",
shape_by = "condition",
n_pc = 20)
plot(compare_figure$p_umap)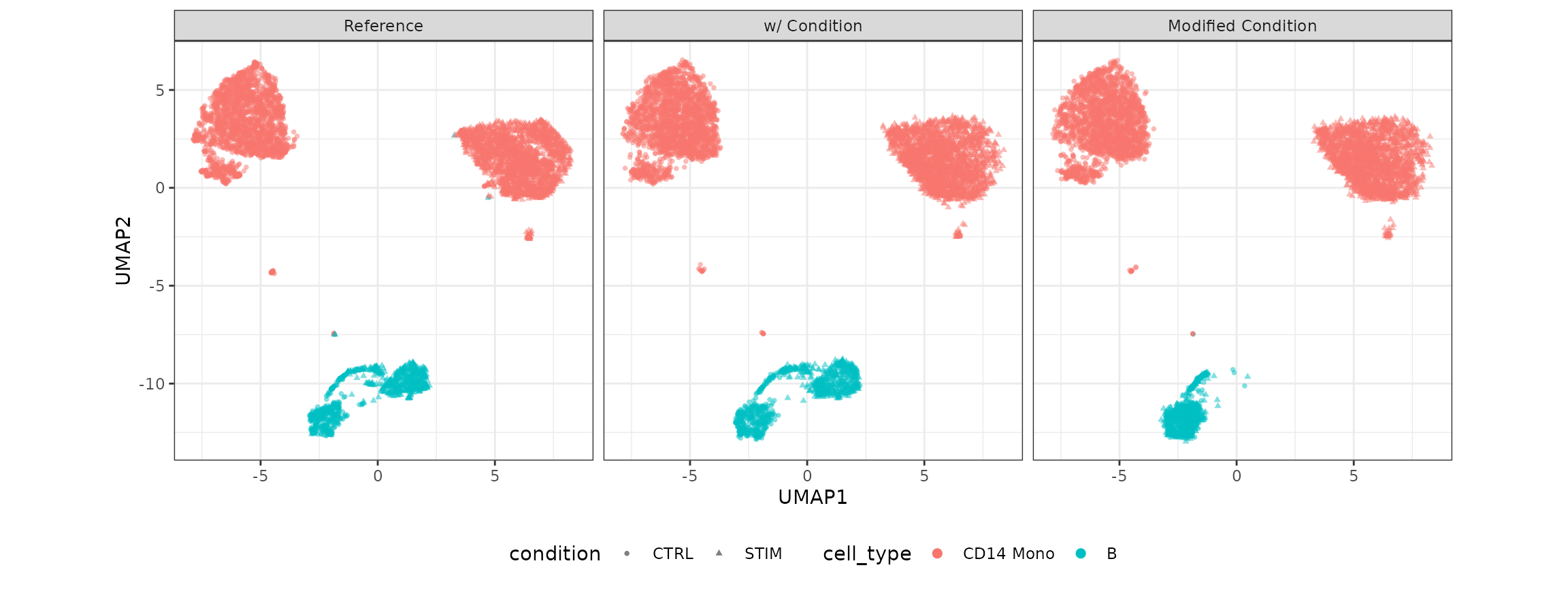
Session information
sessionInfo()
#> R version 4.3.1 (2023-06-16)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Ubuntu 20.04.6 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/liblapack.so.3; LAPACK version 3.9.0
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
#> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: America/Los_Angeles
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] ggplot2_3.4.2 SingleCellExperiment_1.22.0
#> [3] SummarizedExperiment_1.30.2 Biobase_2.60.0
#> [5] GenomicRanges_1.52.0 GenomeInfoDb_1.36.1
#> [7] IRanges_2.34.1 S4Vectors_0.38.1
#> [9] BiocGenerics_0.46.0 MatrixGenerics_1.12.2
#> [11] matrixStats_1.0.0 scDesign3_0.99.6
#> [13] BiocStyle_2.28.0
#>
#> loaded via a namespace (and not attached):
#> [1] tidyselect_1.2.0 farver_2.1.1 dplyr_1.1.2
#> [4] bitops_1.0-7 fastmap_1.1.1 RCurl_1.98-1.12
#> [7] digest_0.6.33 lifecycle_1.0.3 survival_3.5-5
#> [10] gamlss.dist_6.0-5 magrittr_2.0.3 compiler_4.3.1
#> [13] rlang_1.1.1 sass_0.4.7 tools_4.3.1
#> [16] utf8_1.2.3 yaml_2.3.7 knitr_1.43
#> [19] askpass_1.1 S4Arrays_1.0.4 labeling_0.4.2
#> [22] mclust_6.0.0 reticulate_1.30 DelayedArray_0.26.6
#> [25] withr_2.5.0 purrr_1.0.1 desc_1.4.2
#> [28] grid_4.3.1 fansi_1.0.4 colorspace_2.1-0
#> [31] scales_1.2.1 MASS_7.3-60 cli_3.6.1
#> [34] mvtnorm_1.2-2 rmarkdown_2.23 crayon_1.5.2
#> [37] ragg_1.2.5 generics_0.1.3 umap_0.2.10.0
#> [40] RSpectra_0.16-1 cachem_1.0.8 stringr_1.5.0
#> [43] zlibbioc_1.46.0 splines_4.3.1 parallel_4.3.1
#> [46] BiocManager_1.30.21.1 XVector_0.40.0 vctrs_0.6.3
#> [49] Matrix_1.6-0 jsonlite_1.8.7 bookdown_0.34
#> [52] gamlss_5.4-12 irlba_2.3.5.1 systemfonts_1.0.4
#> [55] jquerylib_0.1.4 glue_1.6.2 pkgdown_2.0.7
#> [58] stringi_1.7.12 gtable_0.3.3 munsell_0.5.0
#> [61] tibble_3.2.1 pillar_1.9.0 htmltools_0.5.5
#> [64] openssl_2.1.0 gamlss.data_6.0-2 GenomeInfoDbData_1.2.10
#> [67] R6_2.5.1 textshaping_0.3.6 rprojroot_2.0.3
#> [70] evaluate_0.21 lattice_0.21-8 highr_0.10
#> [73] png_0.1-8 memoise_2.0.1 bslib_0.5.0
#> [76] Rcpp_1.0.11 nlme_3.1-162 mgcv_1.9-0
#> [79] xfun_0.39 fs_1.6.3 pkgconfig_2.0.3