Simulate spot-resolution spatial data for cell-type deconvolution
Dongyuan Song
Bioinformatics IDP, University of California, Los Angelesdongyuansong@ucla.edu
Qingyang Wang
Department of Statistics, University of California, Los Angelesqw802@g.ucla.edu
15 September 2023
Source:../../scDesign3/code/vignettes/scDesign3-spatial-deconvolution.Rmd
scDesign3-spatial-deconvolution.Rmd
library(scDesign3)
library(SingleCellExperiment)
library(ggplot2)
library(dplyr)
library(viridis)
library(IOBR)
library(scatterpie)
theme_set(theme_bw())Introduction
In this tutorial, we show how to use scDesign3 to simulate the spot-resolution spatial data, which each spot is a mix of cells from different cell types.
Read in the reference data
The paired scRNA-seq and spatial data were used in CARD. We pre-select the top cell-type marker genes.
MOBSC_sce <- readRDS((url("https://figshare.com/ndownloader/files/40581983")))
MOBSP_sce <- readRDS((url("https://figshare.com/ndownloader/files/40581986")))
print(MOBSC_sce)
#> class: SingleCellExperiment
#> dim: 182 12640
#> metadata(0):
#> assays(2): counts logcounts
#> rownames(182): Grin2b Prkca ... Zic1 Tpi1
#> rowData names(1): rownames.count.
#> colnames(12640): WT1_AAACCTGAGCTGCGAA WT1_AAACCTGGTTTGGCGC ...
#> OC2_TAGTTGGTCGCGATCG OC2_TTGCGTCAGGATCGCA
#> colData names(4): cellType sampleInfo sizeFactor cell_type
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):
print(MOBSP_sce)
#> class: SingleCellExperiment
#> dim: 182 278
#> metadata(0):
#> assays(1): counts
#> rownames(182): Grin2b Prkca ... Zic1 Tpi1
#> rowData names(0):
#> colnames(278): 16.918x16.996 18.017x17.034 ... 25.134x28.934
#> 29.961x18.97
#> colData names(2): spatial1 spatial2
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):Simulation
We first use scDesign3 to estimate the cell-type reference from scRNA-seq data.
set.seed(123)
MOBSC_data <- construct_data(
sce = MOBSC_sce,
assay_use = "counts",
celltype = "cell_type",
pseudotime = NULL,
spatial = NULL,
other_covariates = NULL,
corr_by = "1"
)
MOBSC_marginal <- fit_marginal(
data = MOBSC_data,
predictor = "gene",
mu_formula = "cell_type",
sigma_formula = "cell_type",
family_use = "nb",
n_cores = 2,
usebam = FALSE,
parallelization = "pbmcmapply"
)
MOBSC_copula <- fit_copula(
sce = MOBSC_sce,
assay_use = "counts",
marginal_list = MOBSC_marginal,
family_use = "nb",
copula = "gaussian",
n_cores = 2,
input_data = MOBSC_data$dat
)
MOBSC_para <- extract_para(
sce = MOBSC_sce,
marginal_list = MOBSC_marginal,
n_cores = 2,
family_use = "nb",
new_covariate = MOBSC_data$newCovariate,
data = MOBSC_data$dat
)
MOBSC_newcount <- simu_new(
sce = MOBSC_sce,
mean_mat = MOBSC_para$mean_mat,
sigma_mat = MOBSC_para$sigma_mat,
zero_mat = MOBSC_para$zero_mat,
quantile_mat = NULL,
copula_list = MOBSC_copula$copula_list,
n_cores = 2,
family_use = "nb",
input_data = MOBSC_data$dat,
new_covariate = MOBSC_data$newCovariate,
filtered_gene = MOBSC_data$filtered_gene
)
set.seed(123)
MOBSP_data <- construct_data(
sce = MOBSP_sce,
assay_use = "counts",
celltype = NULL,
pseudotime = NULL,
spatial = c("spatial1", "spatial2"),
other_covariates = NULL,
corr_by = "1"
)
MOBSP_marginal <- fit_marginal(
data = MOBSP_data,
predictor = "gene",
mu_formula = "s(spatial1, spatial2, bs = 'gp', k = 50, m = c(1, 2, 1))",
sigma_formula = "1",
family_use = "nb",
n_cores = 2,
usebam = FALSE,
parallelization = "pbmcmapply"
)
MOBSP_copula <- fit_copula(
sce = MOBSP_sce,
assay_use = "counts",
marginal_list = MOBSP_marginal,
family_use = "nb",
copula = "gaussian",
n_cores = 2,
input_data = MOBSP_data$dat
)
MOBSP_para <- extract_para(
sce = MOBSP_sce,
marginal_list = MOBSP_marginal,
n_cores = 2,
family_use = "nb",
new_covariate = MOBSP_data$newCovariate,
data = MOBSP_data$dat
)Now we get the fitted models for scRNA-seq and spatial data. We need to extract their mean parameters (i.e., expected expression values).
MOBSC_sig_matrix <- sapply(cell_type, function(x) {
rowMeans(t(MOBSC_para$mean_mat)[, colData(MOBSC_sce)$cellType %in% x])
})
MOBSP_matrix <- (t(MOBSP_para$mean_mat))We use CIBERSORT to decompose each spot’s expected expression into cell-type proportions. This step is to set the true cell-type proportions. Please note you can also use other decomposition methods or set the proportion mannully if you have your own design.
sig_matrix <- as.data.frame(MOBSC_sig_matrix)
mixture_file <- as.data.frame(MOBSP_matrix)
proportion_mat <- IOBR::CIBERSORT(sig_matrix, mixture_file, QN = FALSE, absolute = FALSE, perm = 10)
proportion_mat <- proportion_mat[, 1:4]We can visualzie the proportions by pie-chart.
colors_cell_type <- c("#E69F00", "#56B4E9", "#009E73",
"#0072B2")
d_pie <- as_tibble(colData(MOBSP_sce), rownames = "cell") %>% bind_cols(as_tibble(proportion_mat)) %>% dplyr::mutate(region = seq_len(dim(MOBSP_sce)[2])) %>% dplyr::mutate(X= spatial1, Y = spatial2)
p_pie_plot <- ggplot() + geom_scatterpie(aes(x=X, y=Y, group=region), data=d_pie ,
cols = cell_type, color=NA) + coord_fixed(ratio = 1) +
scale_fill_manual(values = colors_cell_type) + coord_equal()+ theme_bw() + theme(legend.position = "left") + theme(
panel.grid.minor = element_blank(),
panel.grid.major = element_blank(),
axis.text.x=element_blank(),
axis.ticks.x=element_blank(),
axis.text.y=element_blank(),
axis.ticks.y=element_blank())+ guides(fill=guide_legend(title="Cell type"))
p_pie_plot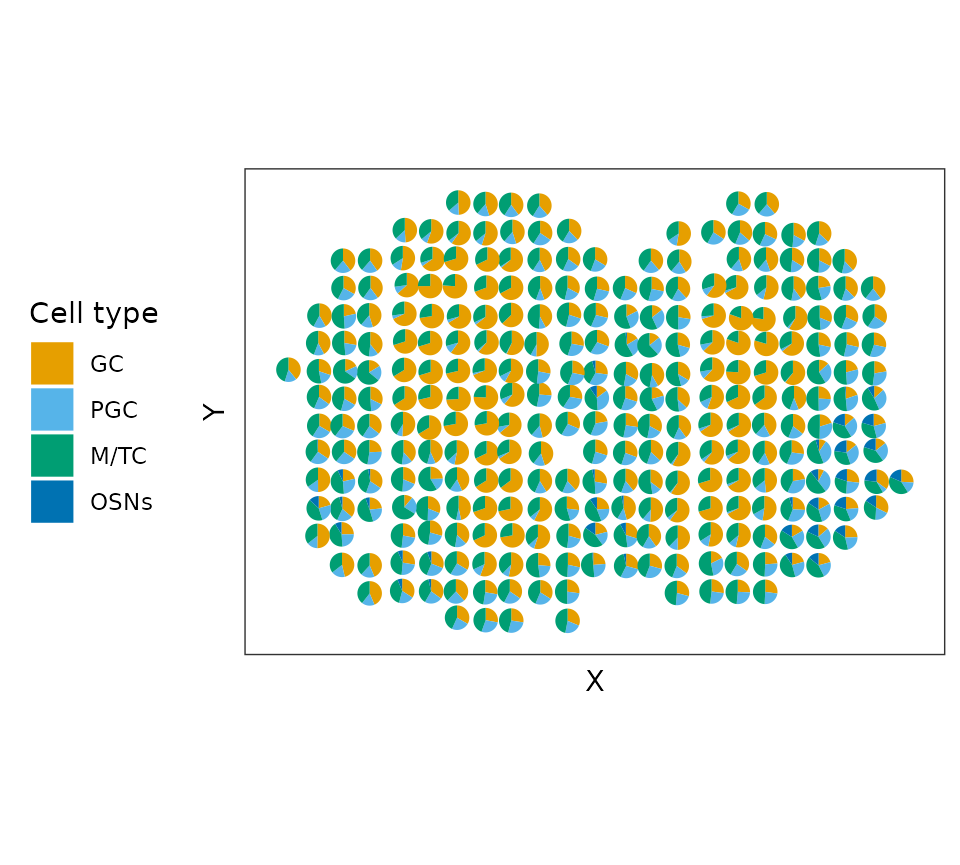
Then we can simulate new spatial data where each spot is the sum of 50 cells/5 (therefore on average 10 cells per spot). Increasing the number of cells will make the spatial data smoother (closer to the expected spatial expression).
set.seed(123)
MOBSCSIM_sce <- MOBSC_sce
counts(MOBSCSIM_sce) <- MOBSC_newcount
MOBSP_new_mixture <- (apply(proportion_mat, 1, function(x) {
n = 50
rowSums(sapply(cell_type, function(y) {
index <- sample(which(colData(MOBSCSIM_sce)$cell_type==y), size = n, replace = FALSE)
rowSums(MOBSC_newcount[, index])*x[y]
}))
}))
MOBSP_new_mixture <- MOBSP_new_mixture/5
### Ceiling to integer
MOBSP_new_mixture <- ceiling(MOBSP_new_mixture)
MOBSPMIX_sce <- MOBSP_sce
counts(MOBSPMIX_sce) <- as.matrix(MOBSP_new_mixture)Finally, we can check the simulated results. We use four cell-type marker genes as the example.
MOBSC_sig_matrix <- sapply(cell_type, function(x) {
rowMeans(t(MOBSC_para$mean_mat)[, colData(MOBSC_sce)$cellType %in% x])
})
MOBSP_sc_mixture <- tcrossprod(as.matrix(MOBSC_sig_matrix), as.matrix(proportion_mat))
rownames(MOBSP_sc_mixture) <- rownames(MOBSP_new_mixture)
location <- colData(MOBSP_sce)
MOBSP_real_tbl <- as_tibble(t(log1p(counts(MOBSP_sce)))) %>% dplyr::mutate(X = location$spatial1,
Y = location$spatial2) %>%
tidyr::pivot_longer(-c("X", "Y"), names_to = "Gene", values_to = "Expression") %>% dplyr::mutate(Method = "Real data")
MOBSP_real_tbl <- transform(MOBSP_real_tbl, Expression=ave(Expression, Gene, FUN=scales::rescale))
MOBSP_mixture_tbl <- as_tibble(t(log1p(MOBSP_new_mixture))) %>% dplyr::mutate(X = location$spatial1,
Y = location$spatial2) %>%
tidyr::pivot_longer(-c("X", "Y"), names_to = "Gene", values_to = "Expression") %>% dplyr::mutate(Method = "scDesign3")
MOBSP_mixture_tbl <- transform(MOBSP_mixture_tbl, Expression=ave(Expression, Gene, FUN=scales::rescale))
MOBSP_tbl <- bind_rows(list(MOBSP_real_tbl, MOBSP_mixture_tbl))
MOBSC_marker <- c("Penk", "Apold1", "Cdhr1", "S100a5")
p_MOB_prop <- MOBSP_tbl %>% dplyr::filter(Gene %in% MOBSC_marker) %>% dplyr::mutate(Gene = factor(Gene, levels = MOBSC_marker)) %>% ggplot(aes(x = X, y = Y, color = Expression)) + ggrastr::rasterize(geom_point(size = 1), dpi = 300) + scale_colour_gradientn(colors = viridis_pal(option = "B", direction = -1)(10), limits=c(0, 1)) + coord_fixed(ratio = 1) + facet_grid(Method ~ Gene ) + theme_bw() + theme(legend.position = "right") + theme(
panel.grid.minor = element_blank(),
panel.grid.major = element_blank(),
axis.text.x=element_blank(),
axis.ticks.x=element_blank(),
axis.text.y=element_blank(),
axis.ticks.y=element_blank())
p_MOB_prop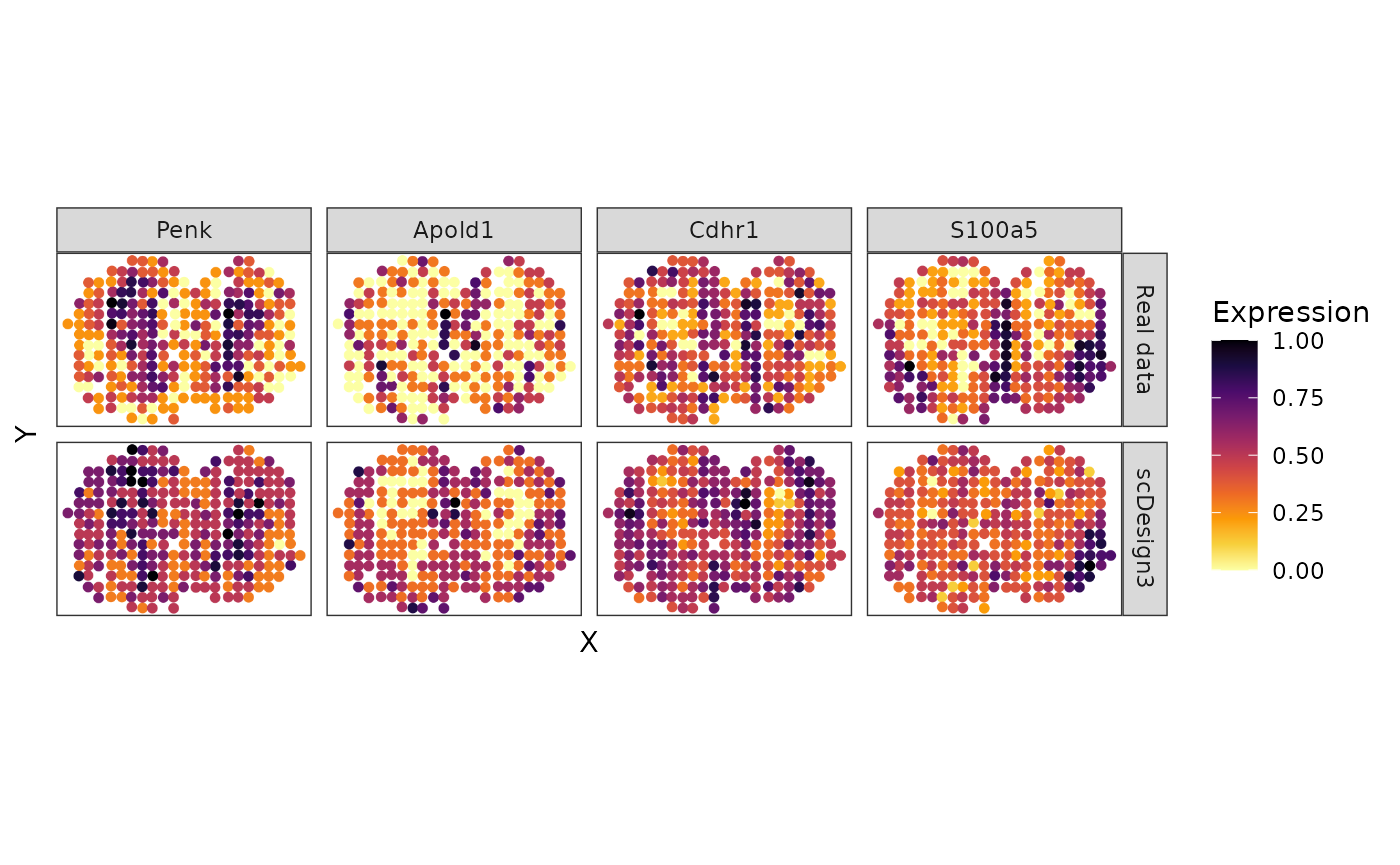
Session information
sessionInfo()
#> R version 4.3.1 (2023-06-16)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Ubuntu 20.04.6 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/liblapack.so.3; LAPACK version 3.9.0
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
#> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: America/Los_Angeles
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] grid stats4 stats graphics grDevices utils datasets
#> [8] methods base
#>
#> other attached packages:
#> [1] scatterpie_0.2.1 IOBR_0.99.9
#> [3] tidyHeatmap_1.8.1 ComplexHeatmap_2.16.0
#> [5] survival_3.5-5 ggpubr_0.6.0
#> [7] tibble_3.2.1 viridis_0.6.4
#> [9] viridisLite_0.4.2 dplyr_1.1.2
#> [11] ggplot2_3.4.2 SingleCellExperiment_1.22.0
#> [13] SummarizedExperiment_1.30.2 Biobase_2.60.0
#> [15] GenomicRanges_1.52.0 GenomeInfoDb_1.36.1
#> [17] IRanges_2.34.1 S4Vectors_0.38.1
#> [19] BiocGenerics_0.46.0 MatrixGenerics_1.12.2
#> [21] matrixStats_1.0.0 scDesign3_0.99.6
#> [23] BiocStyle_2.28.0
#>
#> loaded via a namespace (and not attached):
#> [1] splines_4.3.1 bitops_1.0-7
#> [3] polyclip_1.10-4 preprocessCore_1.62.1
#> [5] graph_1.78.0 XML_3.99-0.14
#> [7] gamlss.data_6.0-2 lifecycle_1.0.3
#> [9] rstatix_0.7.2 pbmcapply_1.5.1
#> [11] doParallel_1.0.17 rprojroot_2.0.3
#> [13] lattice_0.21-8 MASS_7.3-60
#> [15] dendextend_1.17.1 backports_1.4.1
#> [17] magrittr_2.0.3 limma_3.56.2
#> [19] sass_0.4.7 rmarkdown_2.23
#> [21] jquerylib_0.1.4 yaml_2.3.7
#> [23] cowplot_1.1.1 DBI_1.1.3
#> [25] RColorBrewer_1.1-3 abind_1.4-5
#> [27] zlibbioc_1.46.0 quadprog_1.5-8
#> [29] purrr_1.0.1 RCurl_1.98-1.12
#> [31] tweenr_2.0.2 circlize_0.4.15
#> [33] GenomeInfoDbData_1.2.10 KMsurv_0.1-5
#> [35] irlba_2.3.5.1 GSVA_1.48.2
#> [37] annotate_1.78.0 pkgdown_2.0.7
#> [39] DelayedMatrixStats_1.22.1 codetools_0.2-19
#> [41] DelayedArray_0.26.6 ggforce_0.4.1
#> [43] tidyselect_1.2.0 shape_1.4.6
#> [45] farver_2.1.1 ScaledMatrix_1.8.1
#> [47] jsonlite_1.8.7 GetoptLong_1.0.5
#> [49] e1071_1.7-13 iterators_1.0.14
#> [51] systemfonts_1.0.4 foreach_1.5.2
#> [53] tools_4.3.1 ragg_1.2.5
#> [55] Rcpp_1.0.11 glue_1.6.2
#> [57] gridExtra_2.3 mgcv_1.9-0
#> [59] xfun_0.39 DESeq2_1.40.2
#> [61] HDF5Array_1.28.1 withr_2.5.0
#> [63] BiocManager_1.30.21.1 fastmap_1.1.1
#> [65] rhdf5filters_1.12.1 fansi_1.0.4
#> [67] digest_0.6.33 rsvd_1.0.5
#> [69] gamlss_5.4-12 R6_2.5.1
#> [71] textshaping_0.3.6 colorspace_2.1-0
#> [73] Cairo_1.6-0 lpSolve_5.6.18
#> [75] RSQLite_2.3.1 utf8_1.2.3
#> [77] tidyr_1.3.0 generics_0.1.3
#> [79] data.table_1.14.8 class_7.3-22
#> [81] httr_1.4.6 S4Arrays_1.0.4
#> [83] pkgconfig_2.0.3 gtable_0.3.3
#> [85] blob_1.2.4 XVector_0.40.0
#> [87] survMisc_0.5.6 htmltools_0.5.5
#> [89] carData_3.0-5 bookdown_0.34
#> [91] GSEABase_1.62.0 clue_0.3-64
#> [93] scales_1.2.1 tidyverse_2.0.0
#> [95] png_0.1-8 corrplot_0.92
#> [97] ggfun_0.1.1 knitr_1.43
#> [99] km.ci_0.5-6 rjson_0.2.21
#> [101] nlme_3.1-162 proxy_0.4-27
#> [103] cachem_1.0.8 zoo_1.8-12
#> [105] rhdf5_2.44.0 GlobalOptions_0.1.2
#> [107] stringr_1.5.0 vipor_0.4.5
#> [109] parallel_4.3.1 AnnotationDbi_1.62.2
#> [111] ggrastr_1.0.2 desc_1.4.2
#> [113] pillar_1.9.0 vctrs_0.6.3
#> [115] car_3.1-2 BiocSingular_1.16.0
#> [117] beachmat_2.16.0 xtable_1.8-4
#> [119] cluster_2.1.4 beeswarm_0.4.0
#> [121] gamlss.dist_6.0-5 evaluate_0.21
#> [123] mvtnorm_1.2-2 cli_3.6.1
#> [125] locfit_1.5-9.8 compiler_4.3.1
#> [127] rlang_1.1.1 crayon_1.5.2
#> [129] ggsignif_0.6.4 labeling_0.4.2
#> [131] survminer_0.4.9 mclust_6.0.0
#> [133] ggbeeswarm_0.7.2 fs_1.6.3
#> [135] stringi_1.7.12 BiocParallel_1.34.2
#> [137] munsell_0.5.0 Biostrings_2.68.1
#> [139] limSolve_1.5.6 glmnet_4.1-7
#> [141] Matrix_1.6-0 patchwork_1.1.2
#> [143] sparseMatrixStats_1.12.2 bit64_4.0.5
#> [145] Rhdf5lib_1.22.0 KEGGREST_1.40.0
#> [147] highr_0.10 broom_1.0.5
#> [149] memoise_2.0.1 bslib_0.5.0
#> [151] bit_4.0.5