Perform ClusterDE on a two-domain spatial dataset
Siqi Chen
Computer Science, Central South Universitysiqichen4477@gmail.com
Dongyuan Song
Department of Genetics & Genome Sciences, UConn Healthdongyuansong@ucla.edu
11 December 2025
Source:vignettes/ClusterDE-twodomains.Rmd
ClusterDE-twodomains.RmdDownload data
We selected adjacent layer 6 and WM of 151673 slice from the LIBD Human Dorsolateral Prefrontal Cortex (DLPFC) dataset, which is downloaded in the spatialLIBD R package. We removed the genes expressed in fewer than 20% spots to save computational time here.
# # Download the spot-level data
# spe <- spatialLIBD::fetch_data(type = "spe")
#
# # Select the Layer6 and WM domains in the slice "151673"
# sub_151673 <- spe[, spe$sample_id == "151673"]
# index <- sub_151673$spatialLIBD == "L6" | sub_151673$spatialLIBD == "WM"
# index[which(is.na(index))] <- "NAN"
# sub_151673 <- sub_151673[, index == "TRUE"]
# print(sub_151673)
#
# # Delete the genes that express rate less than 20%
# data <- sub_151673@assays@data$counts
# zero_expre_rate <- apply(data, 1, function(x) {
# zero_true <- x == 0
# zero_num <- length(which(zero_true == TRUE)) / dim(data)[2]
# return(zero_num)
# })
# zero_expre_gene_idx <- which(zero_expre_rate < 0.8)
# sub_151673 <- sub_151673[zero_expre_gene_idx,]
# cat(paste0("The size of data: ", dim(sub_151673)[1], "*", dim(sub_151673)[2], sep = ""))
#
# # Construct the SingleCellExperiment object
# dlpfc_twodomain <- SingleCellExperiment::SingleCellExperiment(list(counts = sub_151673@assays@data$counts))
# # Add colData information of singlecellexperiment
# dlpfc_twodomain$spatial1 <- sub_151673@int_colData@listData$spatialCoords[, 2]
# dlpfc_twodomain$spatial2 <- sub_151673@int_colData@listData$spatialCoords[, 1]
# dlpfc_twodomain$cell_type <- sub_151673@colData$spatialLIBD
# SingleCellExperiment::logcounts(dlpfc_twodomain) <- log1p(SingleCellExperiment::counts(dlpfc_twodomain))
# dlpfc_twodomain <- Seurat::as.Seurat(dlpfc_twodomain)
data(dlpfc_twodomain, package = "ClusterDE")Visualize the real data with two domains (L6 and WM).
# Visualize the real spatial domains
domains <- data.frame(Xaxis = dlpfc_twodomain$spatial1, Yaxis = dlpfc_twodomain$spatial2, Domains = dlpfc_twodomain$cell_type)
#> Loading required package: SeuratObject
#> Loading required package: sp
#>
#> Attaching package: 'SeuratObject'
#> The following objects are masked from 'package:base':
#>
#> intersect, t
ggplot2::ggplot(domains, ggplot2::aes(x = Xaxis, y = Yaxis, col = Domains)) +
ggplot2::geom_point(size = 1.0) +
ggplot2::coord_equal() +
ggplot2::ggtitle("Manual annotation \n (The layer6 and WM in the slice 151673)") +
ggplot2::theme(
plot.title = ggplot2::element_text(size = 10, hjust = 0.5),
panel.grid = ggplot2::element_blank(),
panel.background = ggplot2::element_rect(fill = "gray90"),
panel.border = ggplot2::element_rect(color = "black", fill = NA, linewidth = 0.6),
axis.title.x = ggplot2::element_blank(),
axis.title.y = ggplot2::element_blank(),
axis.ticks.x = ggplot2::element_blank(),
axis.ticks.y = ggplot2::element_blank(),
axis.text.x = ggplot2::element_blank(),
axis.text.y = ggplot2::element_blank()
) +
ggplot2::scale_color_manual(values = c("#5791cc", "#403f92"))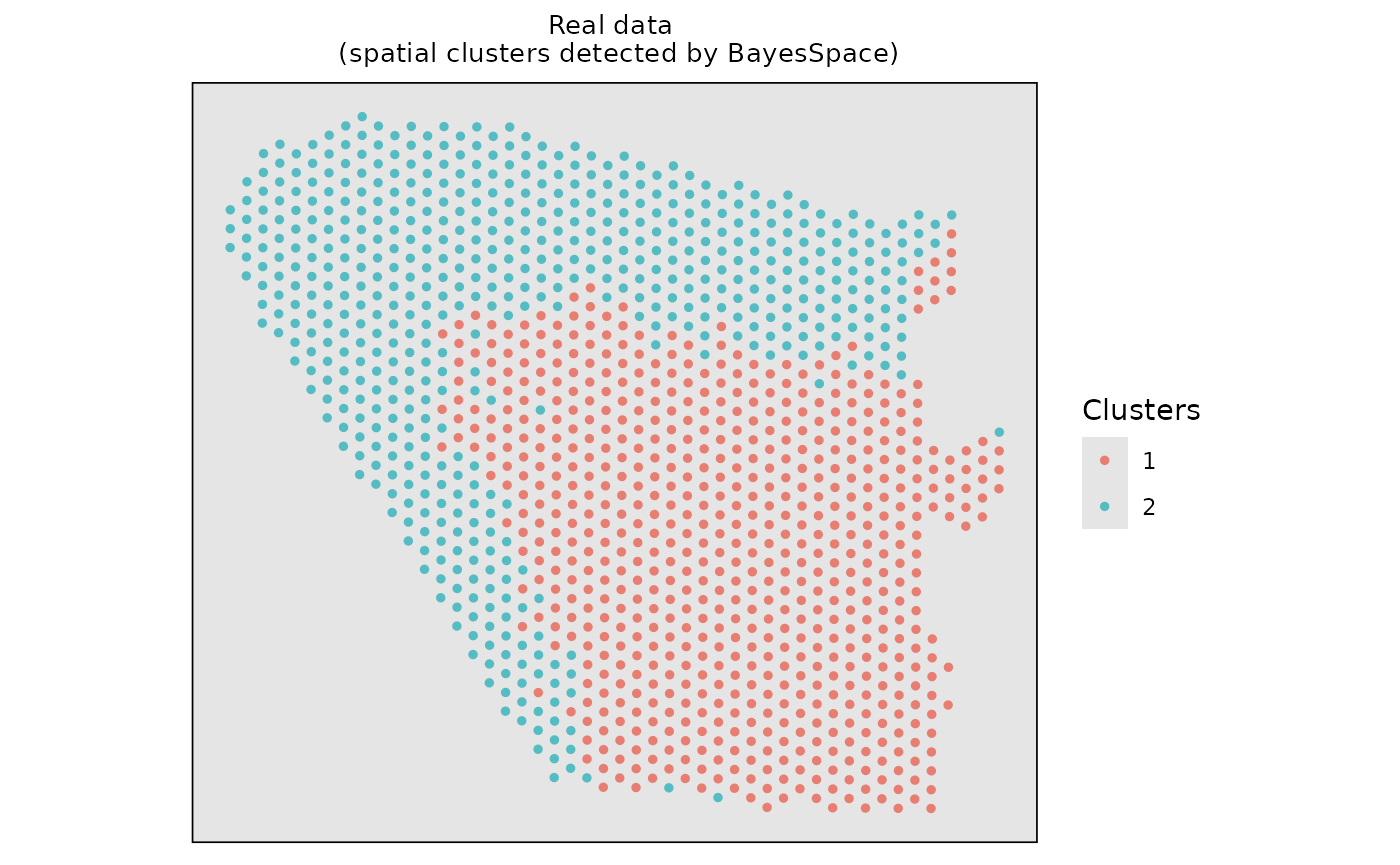
Run the BayesSpace + Seurat pipeline
Firstly, we employed the BayesSpace for spatial clustering. Please note that ClusterDE is designed for 1 vs 1 comparison; therefore, we obtain two spatial clusters for illustration purpose.
# Construct the input of BayesSpace based on real dataset
# The input of BayesSpace is sce object
dlpfc_cluster <- SingleCellExperiment::SingleCellExperiment(
list(counts = Seurat::GetAssayData(dlpfc_twodomain, layer = "counts"))
)
#> Warning: replacing previous import 'S4Arrays::makeNindexFromArrayViewport' by
#> 'DelayedArray::makeNindexFromArrayViewport' when loading 'SummarizedExperiment'
# Add colData information of singlecellexperiment
dlpfc_cluster$row <- dlpfc_twodomain$spatial1
dlpfc_cluster$col <- dlpfc_twodomain$spatial2
# Log-normalize the count data
set.seed(123)
dlpfc_cluster <- BayesSpace::spatialPreprocess(dlpfc_cluster, platform = "ST", n.PCs = 7, log.normalize = T)
# Clustering with BayesSpace
dlpfc_cluster <- BayesSpace::spatialCluster(
dlpfc_cluster,
q = 2,
platform = "ST",
d = 7,
init.method = "mclust",
model = "t",
gamma = 2,
nrep = 1000,
burn.in = 100,
save.chain = T
)
#> Neighbors were identified for 0 out of 1205 spots.
#> Fitting model...
#> You created a large dataset with compression and chunking.
#> The chunk size is equal to the dataset dimensions.
#> If you want to read subsets of the dataset, you should testsmaller chunk sizes to improve read times.
#> You created a large dataset with compression and chunking.
#> The chunk size is equal to the dataset dimensions.
#> If you want to read subsets of the dataset, you should testsmaller chunk sizes to improve read times.
#> Calculating labels using iterations 100 through 1000.Visualize the spatial clustering results based on the real data.
# Visualize the spatial cluster
clusters <- data.frame(Xaxis = dlpfc_cluster$row, Yaxis = dlpfc_cluster$col, Clusters = as.character(dlpfc_cluster$spatial.cluster))
ggplot2::ggplot(clusters, ggplot2::aes(x = Xaxis, y = Yaxis, col = Clusters)) +
ggplot2::geom_point(size = 1.0) +
ggplot2::coord_equal() +
ggplot2::ggtitle("Real data \n (spatial clusters detected by BayesSpace)") +
ggplot2::theme(
plot.title = ggplot2::element_text(size = 10, hjust = 0.5),
panel.grid = ggplot2::element_blank(),
panel.background = ggplot2::element_rect(fill = "gray90"),
panel.border = ggplot2::element_rect(color = "black", fill = NA, size = 0.6),
axis.title.x = ggplot2::element_blank(),
axis.title.y = ggplot2::element_blank(),
axis.ticks.x = ggplot2::element_blank(),
axis.ticks.y = ggplot2::element_blank(),
axis.text.x = ggplot2::element_blank(),
axis.text.y = ggplot2::element_blank()
) +
ggplot2::scale_color_manual(values = c("#e87d72", "#54bcc2"))
#> Warning: The `size` argument of `element_rect()` is deprecated as of ggplot2 3.4.0.
#> ℹ Please use the `linewidth` argument instead.
#> This warning is displayed once every 8 hours.
#> Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
#> generated.
Then, we used the common DE method (Wilcoxon Rank Sum Test) to identify domain marker genes between the two spatial clusters.
# Identify domain marker genes in the real dataset based on the BayesSpace clustering result, follow Seurat tutorial
dlpfc_twodomain <- Seurat::as.Seurat(dlpfc_cluster)
#> Warning: Keys should be one or more alphanumeric characters followed by an
#> underscore, setting key from PC to PC_
Seurat::Idents(dlpfc_twodomain) <- "spatial.cluster"
original_markers <- Seurat::FindMarkers(
object = dlpfc_twodomain,
ident.1 = 2,
ident.2 = 1,
test.use = "wilcox",
logfc.threshold = 0,
min.pct = 0,
min.cells.feature = 1,
min.cells.group = 1
)
original_markers <- original_markers[original_markers$avg_log2FC > 0,]
head(original_markers)
#> p_val avg_log2FC pct.1 pct.2 p_val_adj
#> ENSG00000198804 6.479948e-127 0.9043609 1.000 1.000 1.815033e-123
#> ENSG00000132639 1.894004e-122 1.6959888 0.991 0.926 5.305106e-119
#> ENSG00000198938 1.063328e-108 0.7275062 1.000 1.000 2.978381e-105
#> ENSG00000154146 3.835596e-99 1.4389400 0.978 0.926 1.074350e-95
#> ENSG00000163032 6.974259e-99 1.6499892 0.966 0.821 1.953490e-95
#> ENSG00000187094 6.429586e-96 1.7939567 0.949 0.779 1.800927e-92Find DEGs using ClusterDE
We can use findMarkers() from ClusterDE with spatial specified as name of X and Y coordinates in the data meta columns. Here in the example, coordinates are stored in row and col.
res <- ClusterDE::findMarkers(dlpfc_twodomain, ident.1 = 2, ident.2 = 1, spatial = c("row", "col"))
#> 100% of genes are used in correlation modelling.
#> 0/1: Neighbors were identified for 0 out of 1205 spots.
#> 0/1: Fitting model...
#> 0/1: You created a large dataset with compression and chunking.
#> The chunk size is equal to the dataset dimensions.
#> If you want to read subsets of the dataset, you should testsmaller chunk sizes to improve read times.
#> 0/1: You created a large dataset with compression and chunking.
#> The chunk size is equal to the dataset dimensions.
#> If you want to read subsets of the dataset, you should testsmaller chunk sizes to improve read times.
#> 0/1: Calculating labels using iterations 100 through 1000.
#> 0/1: Normalizing layer: counts
head(res)
#> # A tibble: 6 × 3
#> gene cs record
#> <chr> <dbl> <dbl>
#> 1 ENSG00000132639 53.9 1
#> 2 ENSG00000171617 47.9 1
#> 3 ENSG00000163032 46.2 1
#> 4 ENSG00000198804 41.2 1
#> 5 ENSG00000139970 38.1 1
#> 6 ENSG00000128656 37.2 1Visualize top marker genes
We visualize the top DE genes from ClusterDE. As expected, the top genes detected by ClusterDE exhibit clear spatial expression patterns.
top_genes_clusterde <- res$gene[1:6]
expr <- Seurat::GetAssayData(dlpfc_twodomain, layer = "data")[top_genes_clusterde, , drop = F]
x_coord <- dlpfc_twodomain@meta.data$row
y_coord <- dlpfc_twodomain@meta.data$col
plots <- lapply(top_genes_clusterde, function(gene) {
df <- data.frame(
X = x_coord,
Y = y_coord,
Expr = as.numeric(expr[gene,])
)
ggplot2::ggplot(df, ggplot2::aes(x = X, y = Y, color = Expr)) +
ggplot2::geom_point(size = 1) +
ggplot2::scale_colour_gradientn(colors = viridis::viridis_pal(option = "magma")(10)) +
ggplot2::coord_fixed() +
ggplot2::ggtitle(gene) +
ggplot2::theme_void() +
ggplot2::theme(legend.position = "bottom")
})
patchwork::wrap_plots(plots, nrow = 2)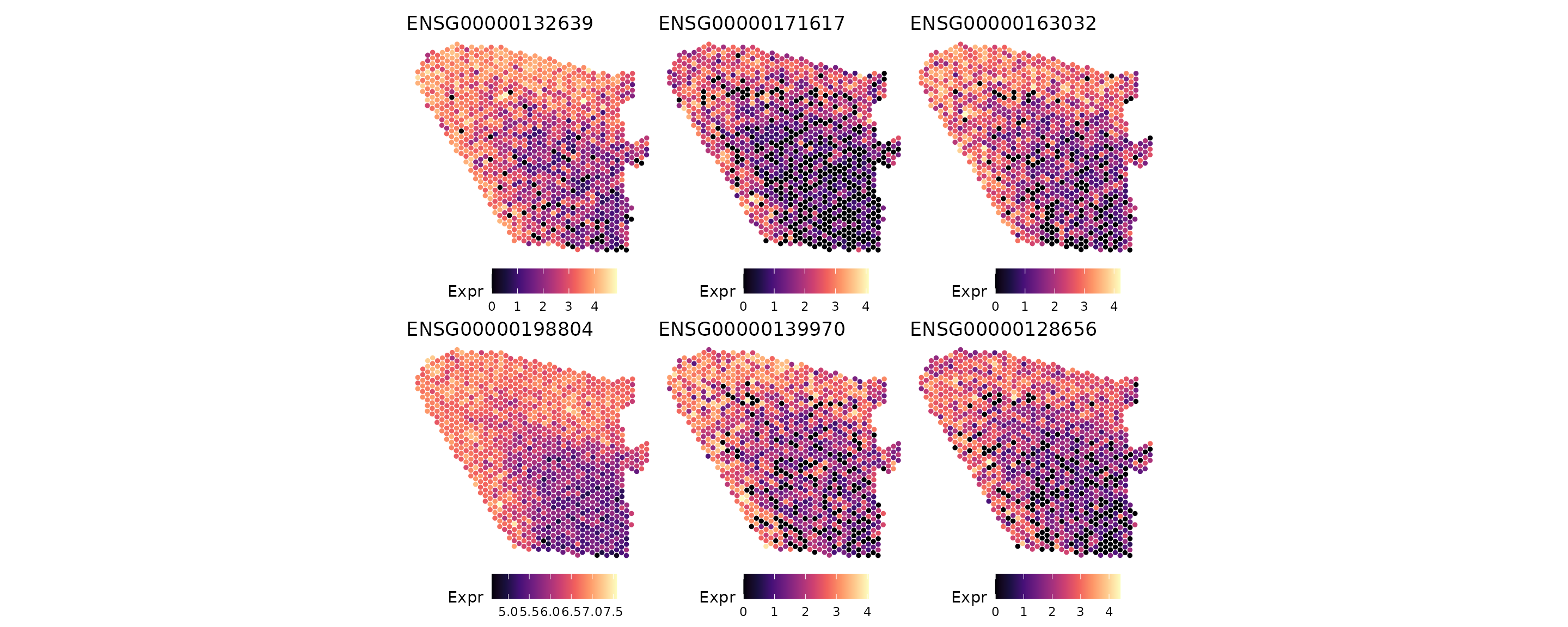
Session information
sessionInfo()
#> R version 4.3.1 (2023-06-16)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Red Hat Enterprise Linux 8.10 (Ootpa)
#>
#> Matrix products: default
#> BLAS: /sw/pkgs/arc/stacks/gcc/10.3.0/R/4.3.1/lib64/R/lib/libRblas.so
#> LAPACK: /sw/pkgs/arc/stacks/gcc/10.3.0/R/4.3.1/lib64/R/lib/libRlapack.so; LAPACK version 3.11.0
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
#> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: America/Detroit
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] SeuratObject_5.2.0 sp_2.2-0 BiocStyle_2.30.0
#>
#> loaded via a namespace (and not attached):
#> [1] fs_1.6.6 matrixStats_1.5.0
#> [3] spatstat.sparse_3.1-0 bitops_1.0-9
#> [5] httr_1.4.7 RColorBrewer_1.1-3
#> [7] backports_1.5.0 tools_4.3.1
#> [9] sctransform_0.4.2 utf8_1.2.6
#> [11] R6_2.6.1 DirichletReg_0.7-2
#> [13] lazyeval_0.2.2 uwot_0.2.3
#> [15] rhdf5filters_1.14.1 withr_3.0.2
#> [17] ClusterDE_0.99.4 gridExtra_2.3
#> [19] progressr_0.17.0 cli_3.6.5
#> [21] Biobase_2.62.0 textshaping_1.0.4
#> [23] spatstat.explore_3.5-3 fastDummies_1.7.5
#> [25] sandwich_3.1-1 labeling_0.4.3
#> [27] sass_0.4.10 Seurat_5.3.1
#> [29] S7_0.2.0 spatstat.data_3.1-9
#> [31] ggridges_0.5.7 pbapply_1.7-4
#> [33] pkgdown_2.2.0 systemfonts_1.3.1
#> [35] scater_1.30.1 parallelly_1.45.1
#> [37] limma_3.58.1 RSQLite_2.4.5
#> [39] generics_0.1.4 ica_1.0-3
#> [41] spatstat.random_3.4-2 dplyr_1.1.4
#> [43] Matrix_1.6-5 ggbeeswarm_0.7.3
#> [45] S4Vectors_0.40.2 abind_1.4-8
#> [47] lifecycle_1.0.4 yaml_2.3.10
#> [49] edgeR_4.0.16 SummarizedExperiment_1.32.0
#> [51] rhdf5_2.46.1 SparseArray_1.2.4
#> [53] BiocFileCache_2.10.2 Rtsne_0.17
#> [55] grid_4.3.1 blob_1.2.4
#> [57] promises_1.4.0 dqrng_0.4.1
#> [59] crayon_1.5.3 miniUI_0.1.2
#> [61] lattice_0.21-8 beachmat_2.18.1
#> [63] cowplot_1.2.0 kde1d_1.1.1
#> [65] pillar_1.11.1 knitr_1.50
#> [67] metapod_1.10.1 GenomicRanges_1.54.1
#> [69] randtoolbox_2.0.5 xgboost_3.1.2.1
#> [71] future.apply_1.20.0 codetools_0.2-19
#> [73] glue_1.8.0 spatstat.univar_3.1-4
#> [75] data.table_1.17.8 vctrs_0.6.5
#> [77] png_0.1-8 spam_2.11-1
#> [79] gtable_0.3.6 assertthat_0.2.1
#> [81] cachem_1.1.0 xfun_0.53
#> [83] S4Arrays_1.2.1 mime_0.13
#> [85] coop_0.6-3 rngWELL_0.10-10
#> [87] coda_0.19-4.1 survival_3.5-5
#> [89] SingleCellExperiment_1.24.0 maxLik_1.5-2.1
#> [91] statmod_1.5.1 bluster_1.12.0
#> [93] BayesSpace_1.12.0 fitdistrplus_1.2-4
#> [95] ROCR_1.0-11 bettermc_1.2.2.9000
#> [97] nlme_3.1-162 bit64_4.6.0-1
#> [99] filelock_1.0.3 RcppAnnoy_0.0.22
#> [101] GenomeInfoDb_1.38.8 bslib_0.9.0
#> [103] irlba_2.3.5.1 vipor_0.4.7
#> [105] KernSmooth_2.23-21 otel_0.2.0
#> [107] BiocGenerics_0.48.1 DBI_1.2.3
#> [109] tidyselect_1.2.1 bit_4.6.0
#> [111] compiler_4.3.1 curl_7.0.0
#> [113] BiocNeighbors_1.20.2 desc_1.4.3
#> [115] DelayedArray_0.28.0 plotly_4.11.0
#> [117] bookdown_0.45 checkmate_2.3.3
#> [119] scales_1.4.0 lmtest_0.9-40
#> [121] mvnfast_0.2.8 rvinecopulib_0.7.3.1.0
#> [123] stringr_1.5.2 digest_0.6.37
#> [125] goftest_1.2-3 presto_1.0.0
#> [127] spatstat.utils_3.2-0 rmarkdown_2.30
#> [129] XVector_0.42.0 htmltools_0.5.8.1
#> [131] pkgconfig_2.0.3 sparseMatrixStats_1.14.0
#> [133] MatrixGenerics_1.14.0 dbplyr_2.5.1
#> [135] fastmap_1.2.0 rlang_1.1.6
#> [137] htmlwidgets_1.6.4 shiny_1.11.1
#> [139] DelayedMatrixStats_1.24.0 farver_2.1.2
#> [141] jquerylib_0.1.4 zoo_1.8-14
#> [143] jsonlite_2.0.0 BiocParallel_1.36.0
#> [145] mclust_6.1.1 BiocSingular_1.18.0
#> [147] RCurl_1.98-1.17 magrittr_2.0.4
#> [149] Formula_1.2-5 scuttle_1.12.0
#> [151] GenomeInfoDbData_1.2.11 dotCall64_1.2
#> [153] patchwork_1.3.2 Rhdf5lib_1.24.2
#> [155] Rcpp_1.1.0 viridis_0.6.5
#> [157] reticulate_1.44.0 stringi_1.8.7
#> [159] zlibbioc_1.48.2 MASS_7.3-60
#> [161] gamlss.dist_6.1-1 plyr_1.8.9
#> [163] parallel_4.3.1 listenv_0.9.1
#> [165] ggrepel_0.9.6 deldir_2.0-4
#> [167] splines_4.3.1 tensor_1.5.1
#> [169] locfit_1.5-9.12 igraph_2.2.1
#> [171] spatstat.geom_3.6-0 RcppHNSW_0.6.0
#> [173] reshape2_1.4.4 stats4_4.3.1
#> [175] ScaledMatrix_1.10.0 evaluate_1.0.5
#> [177] scran_1.30.2 BiocManager_1.30.26
#> [179] httpuv_1.6.16 miscTools_0.6-28
#> [181] RANN_2.6.2 tidyr_1.3.1
#> [183] purrr_1.1.0 polyclip_1.10-7
#> [185] future_1.67.0 scattermore_1.2
#> [187] ggplot2_4.0.0 rsvd_1.0.5
#> [189] xtable_1.8-4 RSpectra_0.16-2
#> [191] later_1.4.4 viridisLite_0.4.2
#> [193] ragg_1.5.0 tibble_3.3.0
#> [195] memoise_2.0.1 beeswarm_0.4.0
#> [197] IRanges_2.36.0 cluster_2.1.4
#> [199] globals_0.18.0